:format(webp):quality(90)/https://www.descopera.ro/wp-content/uploads/2012/06/9718865/2-cover.jpg)
:format(jpeg):quality(80)/https://www.descopera.ro/wp-content/uploads/2012/06/9718865/1-shutterstock-102810128.jpg)
:format(jpeg):quality(80)/https://www.descopera.ro/wp-content/uploads/2012/06/9718865/3-shutterstock-24228883.jpg)
:format(jpeg):quality(80)/https://www.descopera.ro/wp-content/uploads/2012/06/9718865/4-shutterstock-57995710.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/07/1741781917/1ce61c76b7a7ed31c721fca382c7abc6-t.jpg)
Prionii, spune o definiţie seacă, sunt agenţi infecţioşi alcătuiţi din proteine cu o conformaţie spaţială anormală. Aceste molecule proteice sunt capabile să inducă modificări anormale şi moleculelor proteice sănătoase. Spre deosebire de marea majoritate a agenţilor infecţioşi – virusuri, bacterii, ciuperci, paraziţi – prionii sunt alcătuiţi exclusiv din proteine.
Dar de ce sunt atât de uimitori? Ce anume face din ei nişte ciudăţenii, aşa cum au fost priviţi şi cum încă sunt consideraţi adesea?
De fapt, ei nu sunt ciudăţenii; în natură nimic nu e ciudat, totul a apărut ca rezultat al evoluţiei, un proces cu totul firesc, chiar dacă adesea dificil de descifrat pentru noi. Aşa-zisa ciudăţenie a prionilor se leagă de faptul că ne-au contrazis aşteptările, convingerile; ei sunt diferiţi de ceea ce credeam noi că trebuie să fie un agent patogen, se leagă de o concepţie greşită a oamenilor de ştiinţă (şi consecutiv a publicului larg) care a durat multă vreme. Prionii nu sunt ciudaţi decât pentru că sunt altceva decât eram noi obişnuiţi să vedem cu ochii minţii.
Un agent patogen, un microb, cum le zicem adesea în limbajul de toate zilele, era pentru noi o entitate biologică, un organism, în niciun caz o simplă moleculă de… ceva. Infecţia cu un patogen, gândeam noi, însemna invadarea organismului nostru de către un alt organism, o colonizare de către o formă de viaţă dăunătoare nouă. Pătrunderea unor molecule dăunătoare nu însemna infecţie, ci doar o intoxicare – dăunătoare şi aceea, dar nu molipsitoare, nu dătătoare de molime, nu ceea ce numeam boală infecţioasă.
Şi apoi, au venit prionii şi toate aceste delimitări s-au şters, lucrurile au devenit complicate şi confuze, iar categoriile mentale care ne ajutau să păstrăm în ordine cunoaşterea noastră despre lumea vie au trebuit redefinite.
Descoperirea prionilor, consideră unii oameni de ştiinţă, contrazice ceea ce era cunoscut înainte drept dogma centrală a biologiei moleculare. Enunţată de celebrul Francis Crick (unul dintre laureaţii Premiului Nobel, pentru meritul de a fi descoperit structura ADN-ului, una dintre uriaşele izbânzi ştiinţifice ale biologiei), aceasta afirma că informaţia (înţelegându-se prin aceasta informaţia genetică) nu poate fi transmisă decât într-un singur sens: ADN→ARN→proteine.
Adică: pentru a sintetiza o proteină oarecare, informaţia genetică cuprinsă în anumite gene din ADN este copiată de mARN (ARN mesager), prin procesul de transcripţie şi transportată la structurile care se ocupă de sinteza proteinelor; acolo, citind informaţia adusă de mARN (care reprezintă instrucţiunile după care trebuie construită proteina respectivă, prin asamblarea aminoacizilor componenţi într-o anumită ordine), este sintetizată proteina cu pricina, într-un proces numit translaţie. Din tot ceea ce se ştia despre mecanismele celulare până în anii 1970, nimic nu părea să meargă în lumea vie altfel decât conform acestei scheme de bază, de aceea, conceptul respectiv a şi căpătat numele de dogmă, reflectând oarecum caracterul neclintit pe care savanţii vremii îl atribuiau acestei teorii. Şi dacă asta era dogma, a o pune în discuţie, a încerca să o conteşti, era un fel de erezie. Când, în 1982, savantul american Stanley B. Prusiner a anunţat că reuşise să izoleze agentul encefalopatiei spongiforme transmisibile şi că acesta era o proteină, a avut mult de furcă pentru a convinge o mare parte de comunităţii ştiinţifice, foarte ataşată de dogma centrală a biologiei moleculare.
Dar nimic nu stă pe loc în ştiinţă; în acest domeniu de o vastitate şi o complexitate copleşitoare, care este cercetarea lumii vii, ar trebui să folosim cu prudenţă, mai mult ca oriunde, termeni precum niciodată, absolut, categoric sau dogmă.
Ceea ce părea de neclintit, „adevărul” ştiinţific necontestat cuprins în dogma centrală a biologiei moleculare, a fost zgâlţâit zdravăn odată cu descoperirea unor fenomene care făceau praf ceea ce, până atunci, apăruse tuturor ca fiind în afara oricărei discuţii. Începând cu anul 1970, diverse mecanisme celulare care contraziceau dogma au fost descrise.
Mai întâi, s-a descoperit că informaţia poate merge şi invers, de la ARN spre ADN; adică transcrierea se poate face şi în sens opus, proces numit revers-transcriere şi desfăşurat cu participarea esenţială a unei enzime numite revers-transcriptaza. Cazul cel mai cunoscut este cel al ribovirusurilor, cum ar fi HIV. Aceste virusuri nu au ADN, ci doar ARN, care cuprinde informaţia lor genetică; prin urmare, şi să fi vrut, ca să zicem aşa, n-ar fi avut cum să se conformeze schemei ADN→ARN→proteine. În schimb, s-a descoperit că aceste virusuri fac lucrurile pe dos, adică odată intrate în celule, îşi copiază informaţia lor genetică (din ARN-ul propriu) în ADN-ul celulei-gazdă, cu ajutorul revers-transcriptazei, după care îşi fac de cap, folosind mecanismele moleculare ale gazdei pentru a se multiplica. (Virusurile nu au structură celulară şi nu sunt capabile să se înmulţească singure, ci doar inserându-şi genomul lor în genomul unei celule.) Deci, se poate şi altfel decât zicea dogma centrală a biologiei. (Şi, pentru a fi demonstrat că se poate şi altfel, biologii americani Howard Temin, David Baltimore şi Renato Dulbecco au primit Premiul Nobel pentru Medicină şi Fiziologie, pentru lucrările lor asupra revers-transcriptazei.)
De-a lungul vremii, au mai fost descoperite şi alte procese care contrazic dogma centrală a biologiei moleculare, astfel încât dogma a rămas doar ca un jalon în istoria ştiinţelor biologice, iar savanţii s-au învăţat minte să mai fie atât de categorici când e vorba despre ceva atât de complicat ca mecanismele ce pun în mişcare viaţa.
Una dintre implicaţiile dogmei centrale a biologiei moleculare era convingerea că, pentru a sintetiza o proteină sau a-i modifica structura, ar fi nevoie de un transfer de informaţie genetică, adică de trecerea prin procesul ADN→ARN→proteine. De aceea, descoperirea prionilor, bizarele proteine care modifică alte proteine fără intervenţia niciunui acid nucleic, a fost şi ea o bombă în lumea biologiei. E drept, unii savanţi spun că, din moment ce prionii modifică doar configuraţia spaţială a proteinelor, nu şi secvenţa lor de aminoacizi, ei, tehnic vorbind, n-ar contrazice, de fapt, dogma centrală a biologiei moleculare. O fi, poate lucrurile pot fi privite şi aşa. Dar asta, în fond, e doar o discuţie teoretică. Mult mai important este faptul că, odată cu descoperirea prionilor, s-a descoperit, de fapt, că lumea agenţilor infecţioşi e mult, mult mai complexă decât se credea. Pentru specialişti, faptul că nişte proteine, doar nişte proteine, nişte simple molecule proteice, pot produce infecţii contagioase şi epidemii a fost o lovitură de teatru.
Şi, după constarea că se descoperise ceva ce nici nu se credea că există, a urmat un al doilea şoc, atunci când oamenii de ştiinţă şi-au dat seama ce vătămări produceau aceste nou-descoperite particule infecţioase. Departe de a produce afecţiuni banale, trecătoare ori măcar tratabile, cum sunt foarte multe dintre bolile produse de virusuri, bacterii şi ceilalţi agenţi patogeni „convenţionali”, prionii, din ceea ce ştim până acum despre ei, produc doar boli foarte grave, incurabile şi fatale.
E drept, prionii nu se transmit pe cale aeriană, ca multe dintre virusurile şi bacteriile cunoscute (sau nu ştim încă…), ci prin alte mecanisme. De altfel, mecanismul de transmitere misterios a fost una dintre marile dificultăţile legate de studiul prionilor. Odată înţeles acest mecanism, au putut fi luate măsuri care au limitat foarte mult răspîndirea unor boli prionice.
Bolile de acest tip sunt poduse de mutaţii ale genei PRNP. În starea ei normală, această genă codifică sinteza unei proteine numite PrPC , cu anumite roluri în ţesutul nervos. O mutaţie, o alterare în structura genei, determină sinteza unei proteine anormale, numite PrPSc , care are aceeaşi secvenţă de aminoacizi ca şi proteina normală PrPC (adică este alcătuită din aceleaşi subunităţi, aşezate în aceeaşi ordine), dar are o altă conformaţie spaţială: şirul de aminoacizi care formează molecula este înfăşurat, pliat, încolăcit într-o altă formă decât cea normală. În lumea extraordinar de complicată a proteinelor, configuraţia lor tridimensională este la fel de importantă ca şi secvenţa de aminoacizi: o mică schimbare în aranjamentul spaţial al moleculei şi proteina devine alta, comportându-se diferit. În cazul prionilor, diferenţa este practic, echivalentă cu cea dintre viaţă şi moarte: proteina normală are rostul ei în celulele nervoase, în vreme ce proteina „strâmbă”, mutantă, le distruge.
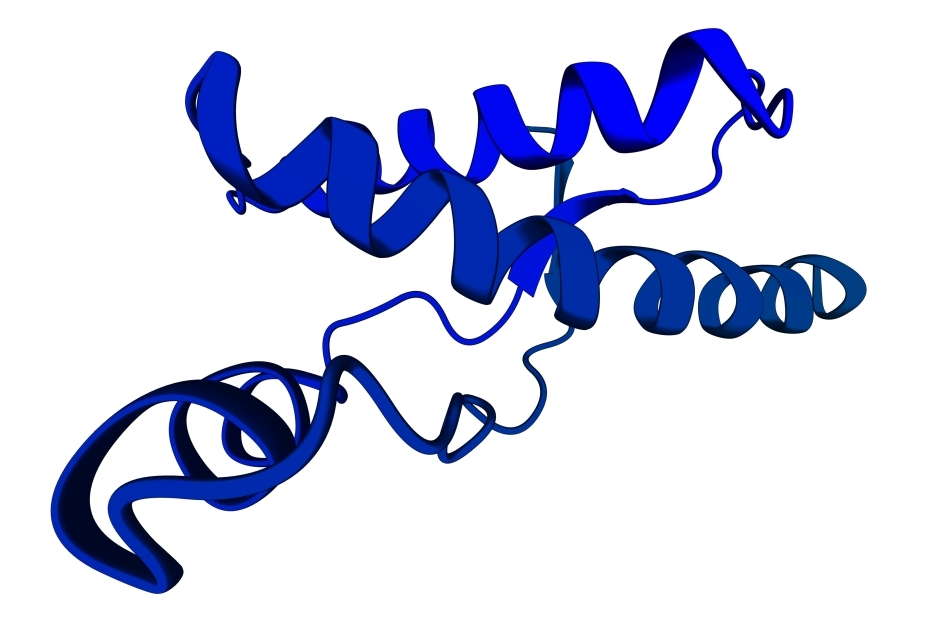
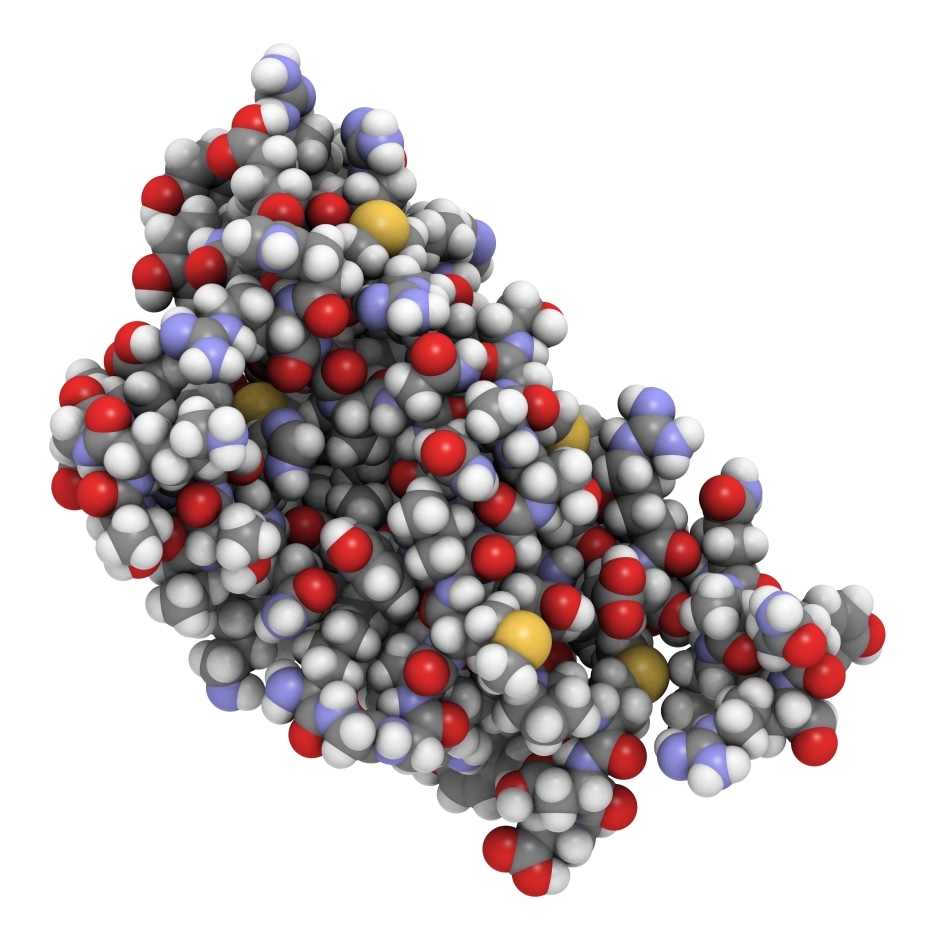
Proteina defectă are înspăimântătoarea capacitate de a „defecta” şi moleculele normale ale proteinei PrPC, făcându-le să adopte configuraţia greşită; este modul în care aceste proteine infecţioase se multiplică. Mecanismele moleculare intime ale acestui proces nu sunt cunoscute în detaliu; cercetări intense, desfăşurate în cele mai performante laboratoare ale lumii, urmăresc elucidarea acestor aspecte, deoarece de cunoaşterea acestor amănunte depinde un lucru extrem de important: modul în care trebuie „proiectate” medicamentele care să combată bolile prionice.
La ora actuală, se cunosc numai câteva maladii produse de prioni. Toate atacă sistemul nervos central, provocând moartea celulelor nervoase, distrugând materia cenuşie, care prezintă ulterior, la autopsie, un aspect caracteristic, părând „ciuruită” de nenumărate orificii şi canalicule, întocmai ca un burete, de unde şi atributul de spongiforme asociat acestor encefalopatii (afecţiuni ale creierului).


La om au fost descrise, de asemenea, un mic număr de encefalopatii spongiforme.
Istoria lor a început cu maladia kuru, observată de savanţii occidentali în anii 1950 la câteva populaţii indigene din Papua Noua Guinee, dar rămasă mult timp o boală misterioasă. Studiile de teren păreau să indice (şi experimentele de laborator au confirmat) că era o boală transmisibilă, însă agentul infecţios nu era cunoscut. În cele din urmă, în anii ’70 s-a descoperit că era vorba despre prioni şi că boala se transmitea datorită unor practici canibale rituale, în cadrul cărora localnicii din unele triburi consumau corpurile unor congeneri decedaţi, pentru a readuce „forţa vitală” a fiinţei în grupul social din care făcuse parte. Printre părţile consumate se număra şi creierul, unde concentraţia de prioni era mare. Se crede că la originea bolii ar fi stat consumul cadavrului unui om care dezvoltase spontan o mutaţie ce dusese la apariţia bolii Creutzfeldt-Jacob (vezi mai jos), cea mai răspândită formă de encefalopatie spongiformă umană.
Boala se manifesta prin debilitate, slăbiciune, incapacitatea de a sta în picioare, accese bruşte de râs şi un tremur puternic şi permanent al întregului corp; în cele din urmă, victima deceda, invariabil.
Impunerea de către australieni (care administrau pe atunci regiunea respectivă din insula Papua Noua Guinee) a unei legi care interzicea practicile canibale în cadrul riturilor funerare, împreună cu eforturile în aceeaşi direcţie ale misiunilor creştine din zonă, au dus la declinul treptat al acestor practici şi, în consecinţă, la scăderea incidenţei bolii kuru. Însă, deoarece are o perioadă lungă de incubaţie (uneori zeci de ani), e încă posibil ca persoane contaminate în urmă cu decenii să dezvolte totuşi boala.
Maladia Creutzfeldt-Jacob (CJD – Creutzfeldt-Jacob Disease) este encefalopatia spongiformă cea mai des întâlnită la om. Asta nu înseamnă că e boală frecventă, dimpotrivă: se înregistrează aproximativ un caz la 1 milion de persoane, în fiecare an.
Au fost descrise trei tipuri:
În cadrul acestui din urmă tip, pe de o parte, au fost descrise forme numite iatrogenice, în care agentul infecţios a fost transmis prin electrozi contaminaţi, prin instrumentar chirugical şi stomatologic incorect sterilizat, prin utilizarea de grefe provenite de la cadavre infectate cu prioni sau prin administrarea de hormon de creştere obţinut din glandele hipofizare ale cadavrelor unor pacienţi cu CJD (acest tip de hormon nu se mai foloseşte azi, iar procedurile standard de sterilizare au devenit mult mai riguroase.)
Pe de altă parte, au fost înregistrate cazuri de contaminare prin consumul anumitor produse alimentare provenite de la vite infectate cu boala vacii nebune. În 1996, în Marea Britanie a fost descrisă aşa-numita variantă (nouă) a CJD, care, se consideră, poate apărea prin contaminare interspecifică (între specii diferite), în cazul respectiv de la vacă (infectată cu boala vacii nebune) la om.
Consumul de măduvă a spinării, de creier de vită şi alte organe interne poate fi riscant, deoarece aceste organe pot conţine prioni; prin urmare, comercializarea acestor produse a fost restricţionată în multe zone; consumul de grăsime, muşchi, lapte, în schimb, nu prezintă riscuri, după opinia specialiştilor.
Boala Creutzfeldt-Jacob se manifestă iniţial prin simptome de demenţă – pierderi de memorie, schimbări de personalitate, tulburări cognitive – şi tulburări ale echilibrului şi sensibilităţii. Apar mişcări necontrolate ale membrelor, apoi stări de slăbiciune care progresează rapid, iar în cele din urmă, coma şi moartea. Boala este invariabil fatală, de obicei decesul survenind în cel mult un an de la apariţia simptomelor. Pentru moment nu există niciun tratament eficient.
În afară de boala Creutzfeldt-Jacob, la om a mai fost descris un număr mic de encefalopatii spongiforme, extrem de rare, din care se cunosc doar câteva zeci de cazuri în întreaga lume: insomnia familială fatală şi sindromul Gerstmann-Sträussler-Scheinker. Caracterizate tot prin afectarea sistemului nervos, din cauza distrugerii neuronilor, şi aceste afecţiuni au o mortalitate de 100%.
Dar medicina progresează neîncetat; chiar dacă, pentru moment, nu există un medicament aprobat pentru tratarea acestor maladii, în experimentele de laborator au fost obţinute unele rezultate pe baza cărora, în viitor, ar putea deveni posibilă corectarea erorilor din configuraţia proteinelor prionice. La urma urmelor, în urmă cu 70 de ani nu dispuneam nici de antibiotice, iar medicamentele antivirale sunt încă şi mai noi. Tratamentele anti-prionice vor fi şi ele disponibile cândva, iar până atunci, prevenirea, inclusiv prin mijloace drastice, a epidemiilor şi a transmiterii de la animale la om, rămâne soluţia cea mai eficace.
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/02/Continentele-Pamantului-ar-fi-mult-mai_shutterstock_descopera-6-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/nasterea-unei-bucati-din-scoarta-oceanica_Royer-et-al_Nature-2026_descopera-1024x578.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/02/Continentele-Pamantului-ar-fi-mult-mai_shutterstock_descopera-6-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/nasterea-unei-bucati-din-scoarta-oceanica_Royer-et-al_Nature-2026_descopera-1024x578.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/07/1589795485/ace10b74564c2e2b135e543a34f8ce5c-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/07/1589795423/b91a901264d9815ba9e58c1192c2a116-t.png)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/07/1766046944/322c463411c50e19225343b5c40746d4-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/07/1687506106/86f4d7625afd6772be3d18d7fef94f26-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/07/1619101788/db50fef16a2def571c4e9665f87685d7-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/07/1604659743/10a72bbdd9dfe1262b27fba502b31bbb-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/07/1741781930/cdf3850b807e74c975fe77129af80e4e-t.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2025/11/primele-stele-ale-Universului_shutterstock_Descopera-4-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/medicament-injectabil-pentru-bronzare_shutterstock_descopera-10-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/mai-intunecate-impulsuri-ale-oamenilor_shutterstock_descopera-10-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/Din-ce-sunt-facuti-atomii_Shutterstock_Descopera-10-1024x683.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/07/1589795438/9425297a3522c846a687867fb8ea85fc-t.png)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2025/03/1589795452/5eb2647afb5148b0ff24692b4fa1d8be-t.jpg)
:format(wepb):quality(80)/https://www.descopera.ro/wp-content/uploads/sfm/2026/07/1593777592/bcdc9a2d059f993634d4a399c74d1711-t.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2024/10/leu-animal-shutter_descopera211221-1024x681.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/nasture-moneda-vikingi-uis_Descopera-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/stresul-1024x573.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/06/Cele-mai-grave-extinctii-ale-Pamantului_shutterstock_descopera-6-1024x517.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2025/11/primele-stele-ale-Universului_shutterstock_Descopera-4-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/medicament-injectabil-pentru-bronzare_shutterstock_descopera-10-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/mai-intunecate-impulsuri-ale-oamenilor_shutterstock_descopera-10-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/Din-ce-sunt-facuti-atomii_Shutterstock_Descopera-10-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/fenomen-vizual-rar_Pushin-et-al_PNAS-2026_descopera-2-1024x494.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/Volodimir-Zelenski-shutter_Descopera-1-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/drone-pentru-ucraina-shutter_Descopera-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/BMW-rusia-auto-shutter_Descopera-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2024/07/constitutia-romania-shutter_descopera-1024x540.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/Taifunul-Bavi-profi_Descopera-1024x683.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/01/Instagram-hackeri-shutter_Descopera-1024x768.jpg)
:format(webp):quality(80)/https://www.descopera.ro/wp-content/uploads/2026/07/Pitbull-concert-shutter_Descopera-1024x683.jpg)